
系統樹ってどうやってつくるの?という初心者向けの記事です。
自己流です。
macOSを使用している方向けです。
間違いがありましたらご指摘よろしくお願いします。
ソフトウェアは系統樹構築に用いられることの多いMEGA11を使用しています。
https://www.megasoftware.net/
フリーソフトウェアですが、解析に十分な機能が備わっています。
追記:2025年2月に関連記事をアップしました。MEGA12では、より直感的な操作と高度な解析が可能になりました。最新の機能や系統樹作成の手順については、こちらの記事をご覧ください。
今回は16SrRNA遺伝子に基づく系統解析を実施していきたいと思います。
ちなみに16SrRNA遺伝子は
- 配列保存性が高い
- 進化速度が遅い
- 1600bp程度と解析に十分な情報量を持つ上、変異しやすいサイトも存在し近縁種間でも比較可
といった特徴をもつことから系統解析によく用いられる遺伝子です。
MEGA11のインストール
OSを選択し、ダウンロードします。
FASTA形式の配列データの用意
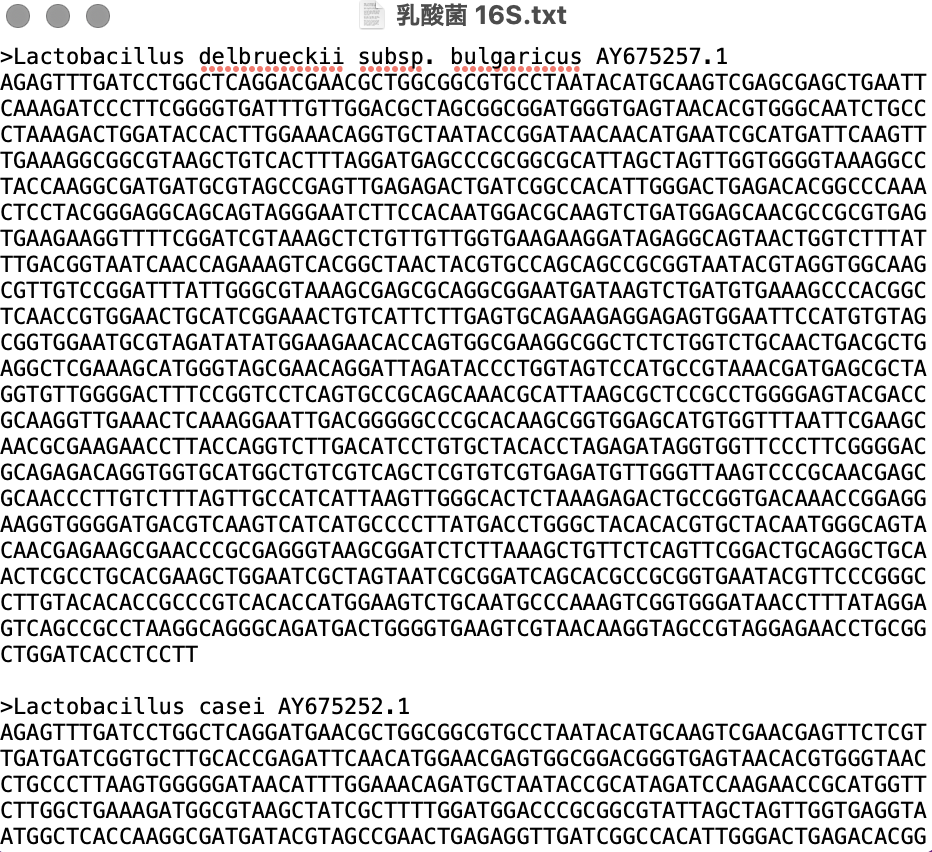
FASTA形式の配列データファイルを用意します。
今回は例として身近な食品に利用されている乳酸菌7種の16SrRNA領域(約1,500bp)の
配列データセットを作成しました。
外群の配列はEscherichia coliを用いました。
| 学名 | NCBI Accession No. | 利用されている食品 |
| Lactobacillus delbrueckii subsp. bulgaricus | AY675257.1 | ヨーグルト |
| Lactobacillus casei | AY675252.1 | ヤクルト |
| Lactobacillus helveticus | FR683085.1 | 乳酸菌飲料 |
| Lactobacillus acidophilus | EF533992.1 | 乳酸菌飲料 |
| Streptococcus thermophilus | AY675258.1 | ヨーグルト |
| Streptococcus salivarius | AY188352.1 | チーズ |
| Lactococcus lactis | AY675242.1 | チーズ |
| Escherichia coli | J01859.1 | なし |
各配列はNCBIのデータベース上で検索しました。https://www.ncbi.nlm.nih.gov/
筆者はmacを使用しているため、テキストエディットでFASTA形式の配列データを作成しています。
MEGA11上で配列アラインメント
MEGA11上でFASTA形式配列データを開く
系統樹を構築する前に配列アラインメントをMEGA11上で実施します。
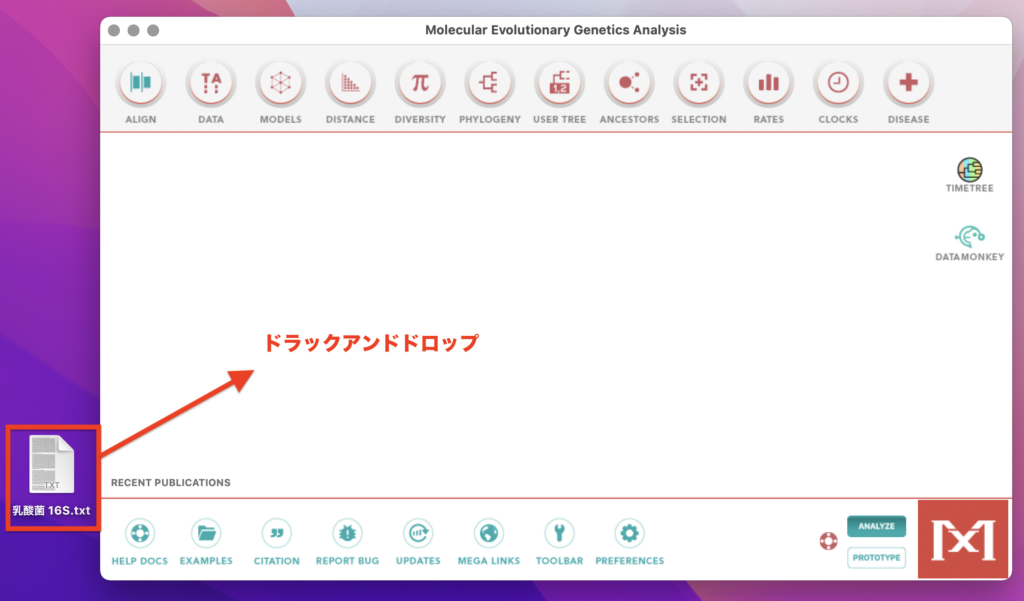
MEGA11を起動し、先ほど作成したFASTA形式のファイルをMEGA11の画面上にドラックアンドドロップします。

ドラックアンドドロップすると、「Text File Editor and Format Converter」なる画面が自動的に起動し、
さきほど作成したファイルの配列データが入力された状態になります。

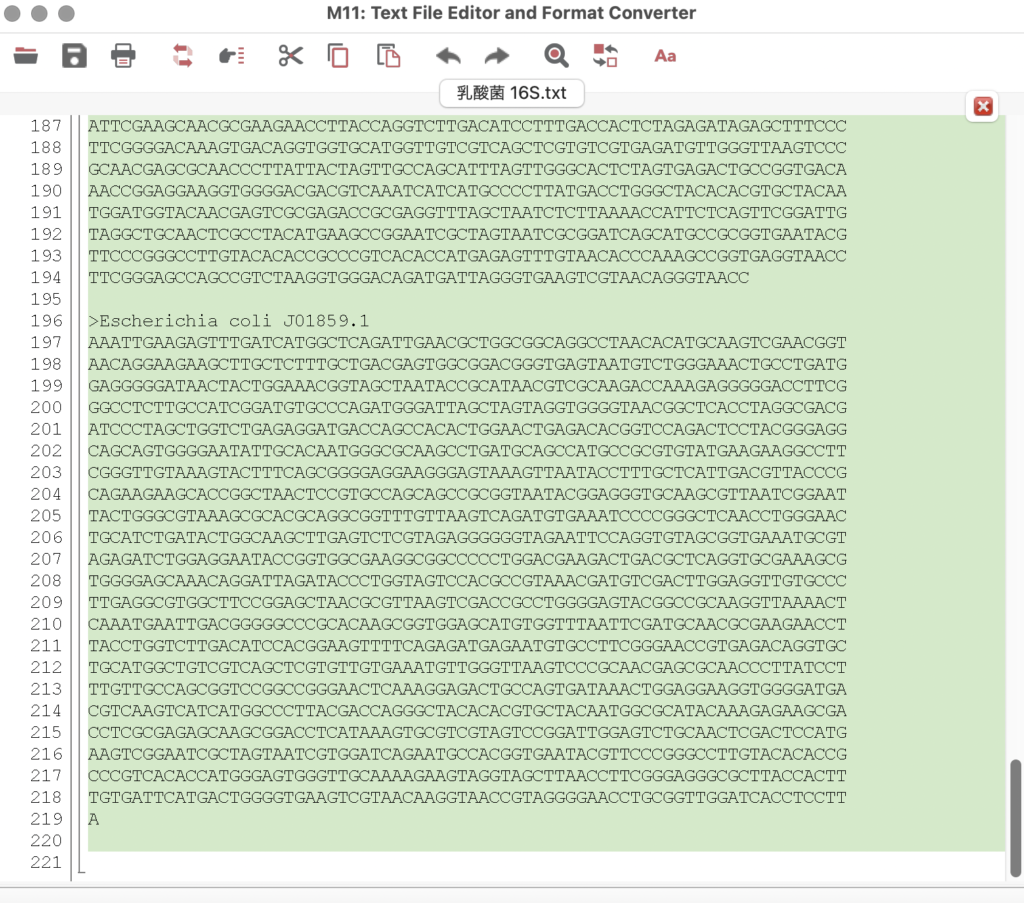
「Edit」→「Select All」で全配列を選択し、コピーします。
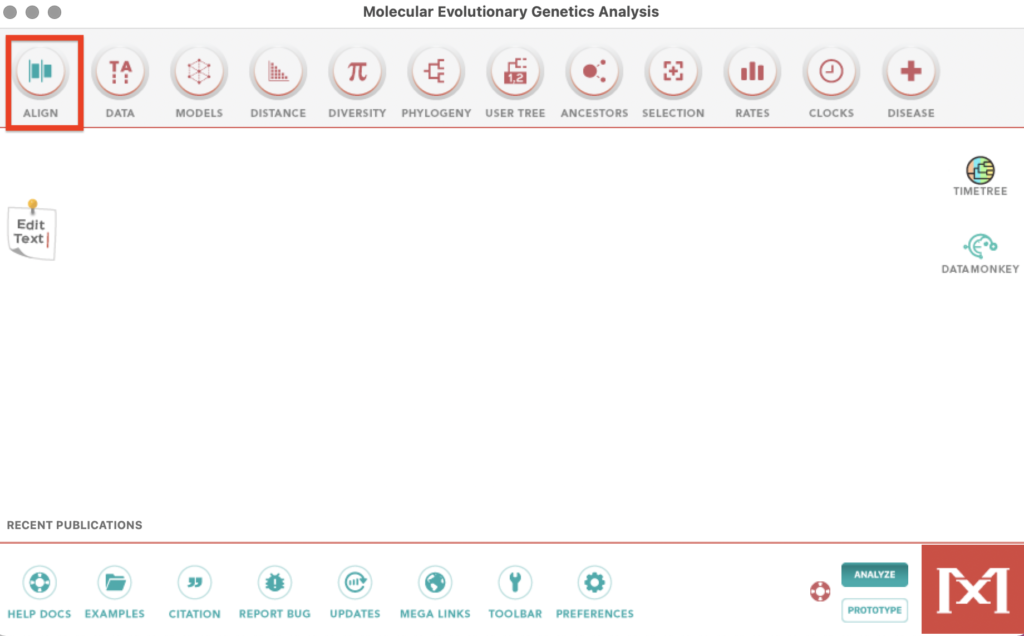
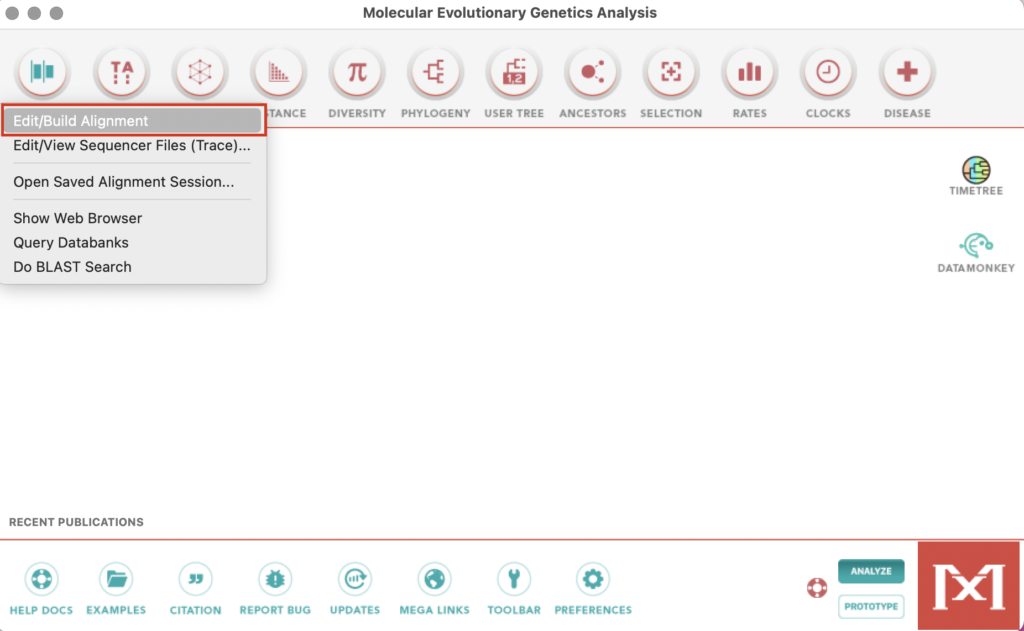
起動時のホーム画面に戻りAlign→Edit/Build Alignmentを選択します。
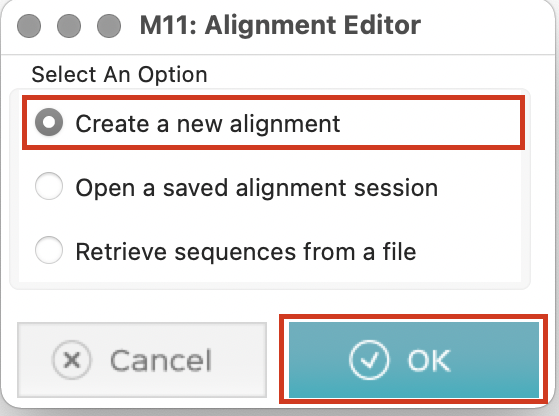
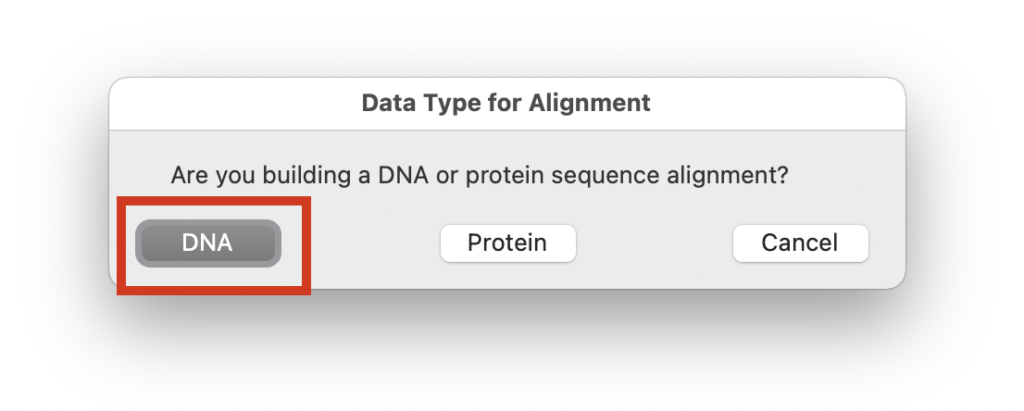
Create a new alignmentを選択し、
「Are you building a DNA or protein sequence alignment?」と聞かれるので、今回はDNAを選択します。
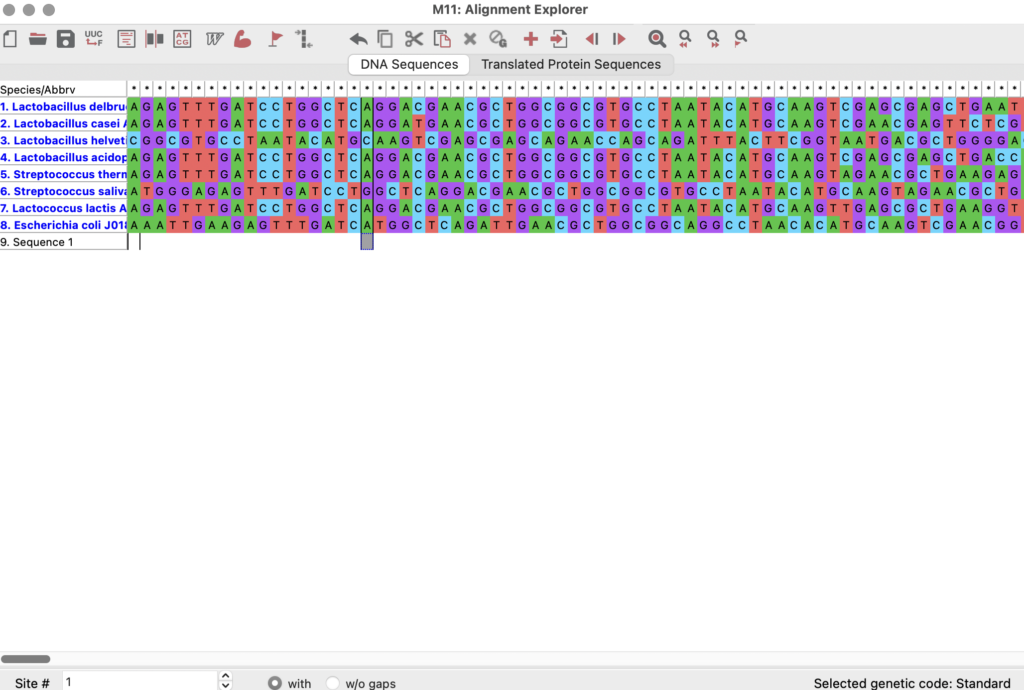
「Alignment Explorer」なる画面が起動するので、先ほどコピーした配列をペーストします。
(下に空白の欄ができた場合は、その行を選択して、上部の「Edit→Select sequence(s)」を選択し、
「Alignment Explorer」上部にある✖️印をクリックして取り除いてください)
ClustalWによるマルチプルアラインメント

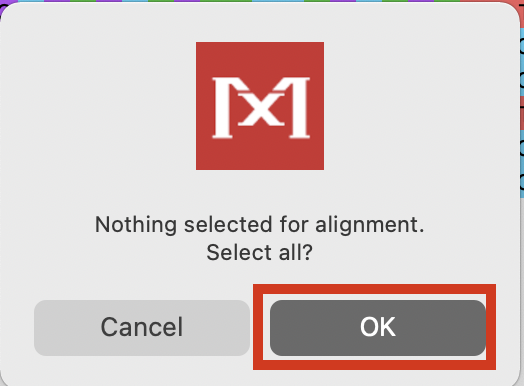
「Alignment Explorer」上部にあるWマークから「Align DNA」を選択します。
今回は全配列をアラインメントの対象とするので、次の画面でOKを押します。
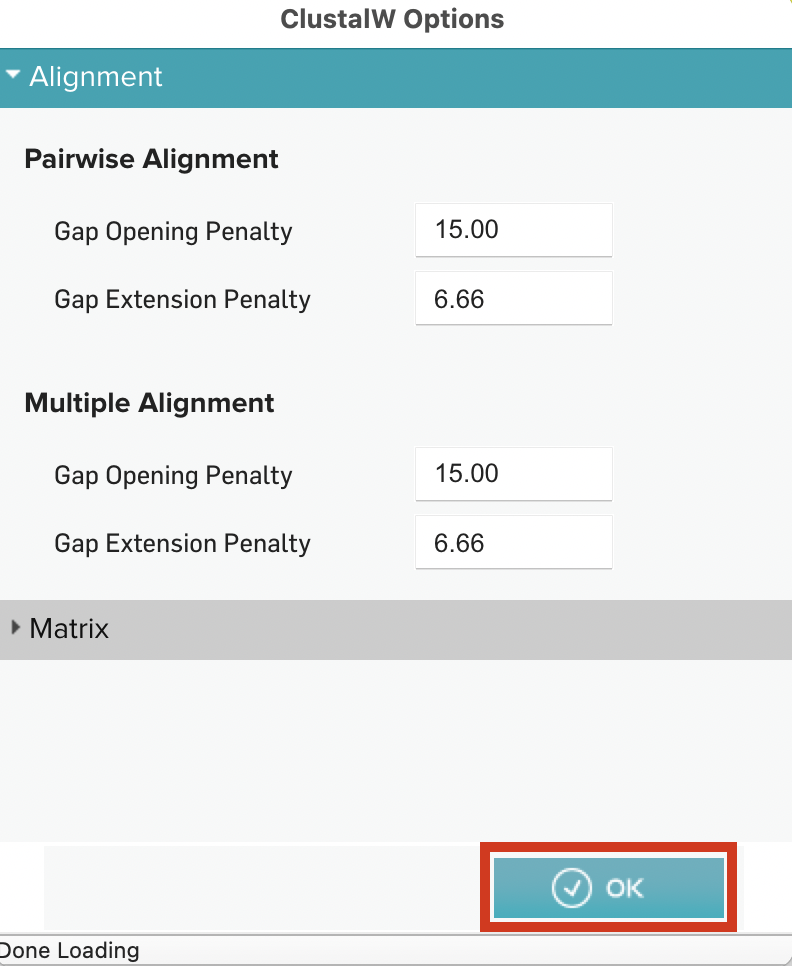
ClustalWのoption画面が表示されるので、今回はデフォルトの設定に従いOKを押してアラインメントを実施しました。

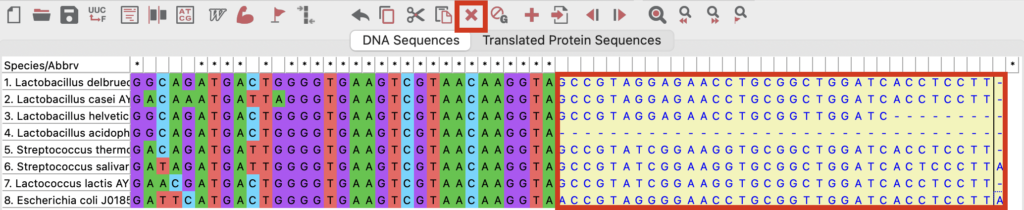
アラインメントが実施されます。
今回はアラインメント前半と後半の配列が揃っていない部分は削除し、
配列が揃っている1422サイトを対象に系統解析を行うことにしました。
両端の配列を削除したのちにもう一度ClustalWによるアラインメントを実施しました。
上部メニューで「Deta」→「Phylogenetic Analysis」を選択します。
これで系統解析に必要なファイル(PhyloAnalysis.meg)が初期画面に表示されていることが確認できます。
(上記の図ではいろんな種類のファイルが生成されている状態ですが、ファイルの数がこれより少なくて問題ありません。)
最適進化モデルの選択
今回は最尤法(maximum likelihood method)に基づく系統樹を構築します。
MEGAでは最尤法の他に近隣結合法、最大節約法、UPGMA法、Minimum-Evolution法(Minimum-Evolution法は距離法の一種のようですが、よくわかっていません)など様々なアルゴリズムに基づく系統樹が構築できるようです。
最尤法においてはいくつかの進化モデルの仮定が存在するため、これから解析を行う配列セットにおける
最適進化モデルは何であるかを判断するための解析を行います。
「MODELS」から「Find Best DNA/Protein Models(ML)」を選択します。
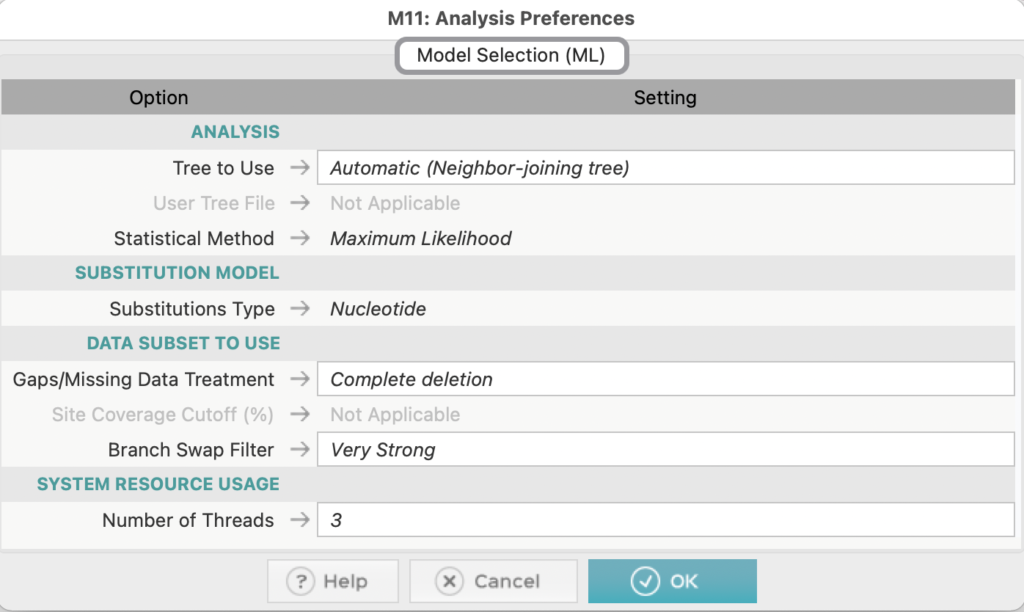
先ほど作成した系統解析用のファイルを選択し、Settingは今回はこのような条件で行いました。
目的に応じ、Settingはカスタマイズしてください。
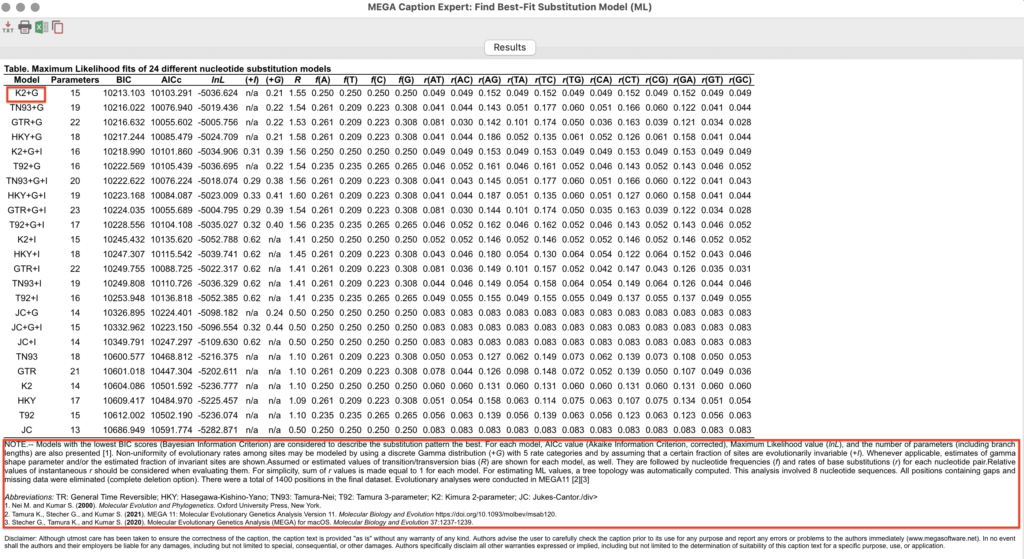
上図のような画面が表示されます。
基本的にこの画面上でいちばん上に表示されたモデル
(今回ならK2+G)が解析した配列セットにおける最適進化モデルであるとなります。
画面下部にそれぞれの指標について説明されています。
+Gは”Non-uniformity of evolutionary rates among sites may be modeled by using a discrete Gamma distribution”を意味し、
K2は”kimura 2-parameter”の略称です。
次の系統樹の構築の際にこの結果をもとにSettingを行います。
系統樹の構築

「PHYLOGENY」から「Construct/Test Maximum Likelihood Tree」を選択します。
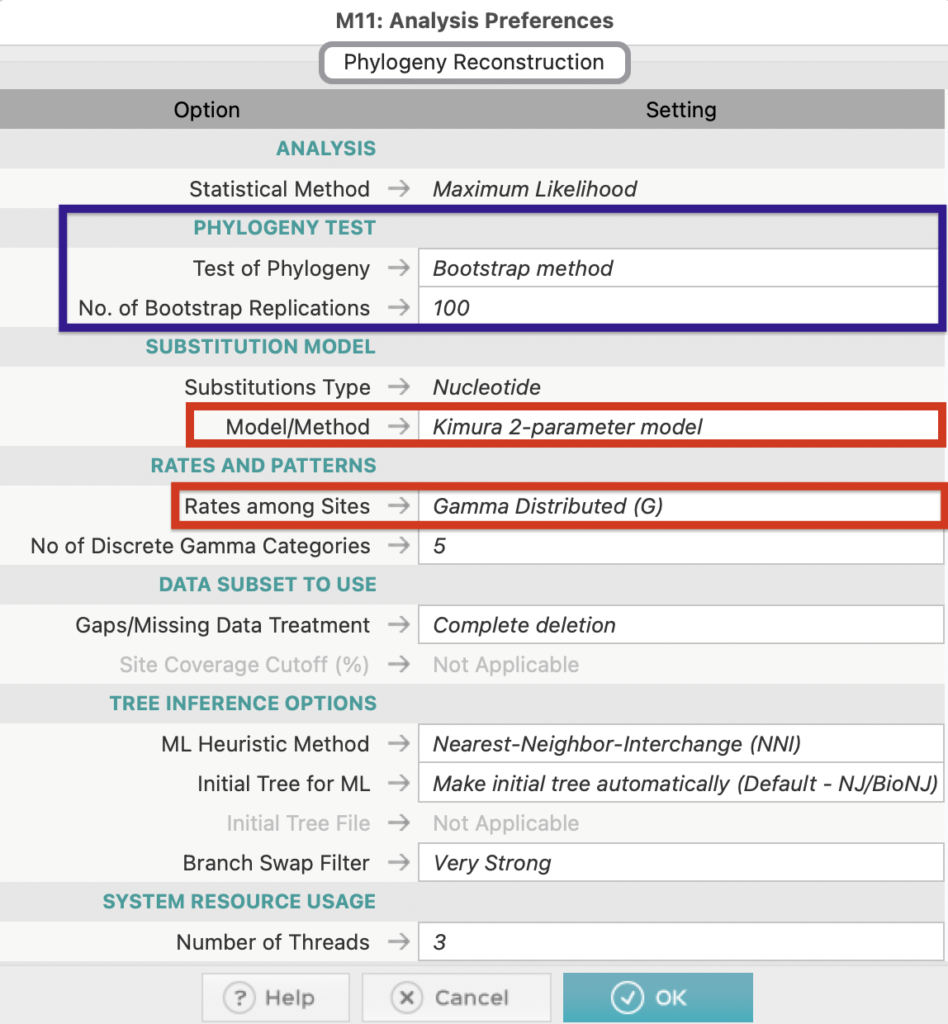
赤四角で示した部分は先ほど確認した最適進化モデルを選択します。
青で囲んだ「PHYLOGENY TEST」は今回は「BootStrap method」を選択し、実施回数は100回としました。
近隣結合法などではBootStrap検定は1000回行うのが一般的ですが、最尤法は計算量が多い解析法であり、
BootStrap検定にも時間がかかるので(実際1000回行うと途中でソフトウェアがダウンすることもあった)
今回は100回に設定しました。
その他のSettingについては今回は上図のように設定しました。
目的に応じてカスタマイズする必要があります。
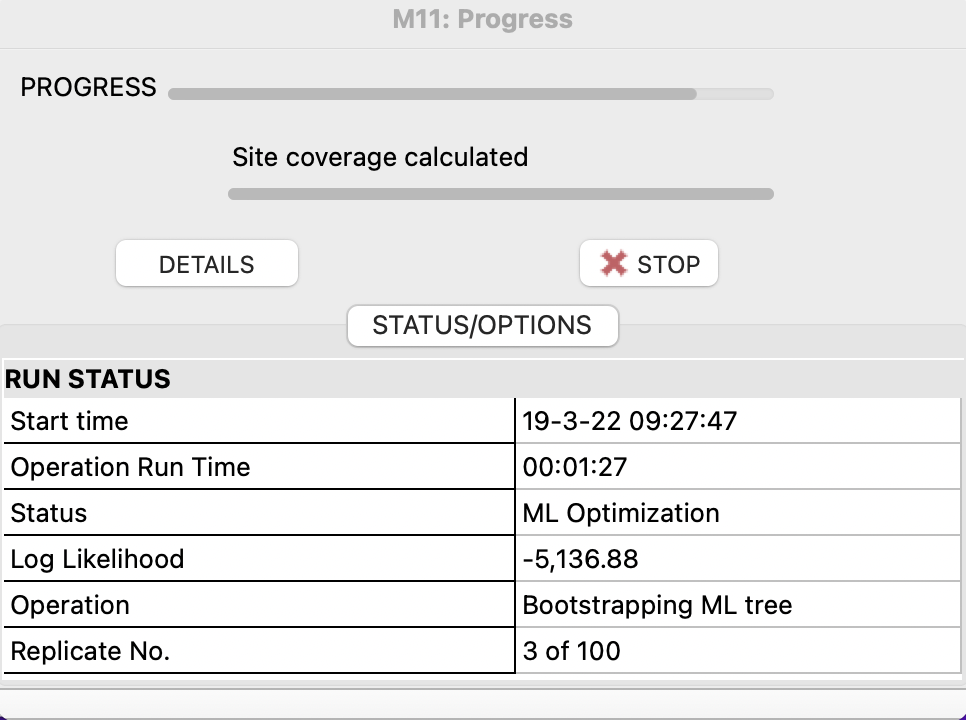
Setting後、OKを押すとこのような画面が出ます。
BootStrap検定が100回終了するまで今回は1時間ほどかかってしまいました。
(今までは5分程度で終了することが多かったのですが…)
設定したパラメーターで系統樹が構築されるか暫定的に確認するために、
まずは「PHYLOGENY TEST」を「none」に設定して系統樹が得られるか確認してから
BootStrap検定をsettingする方が良いかもしれません。
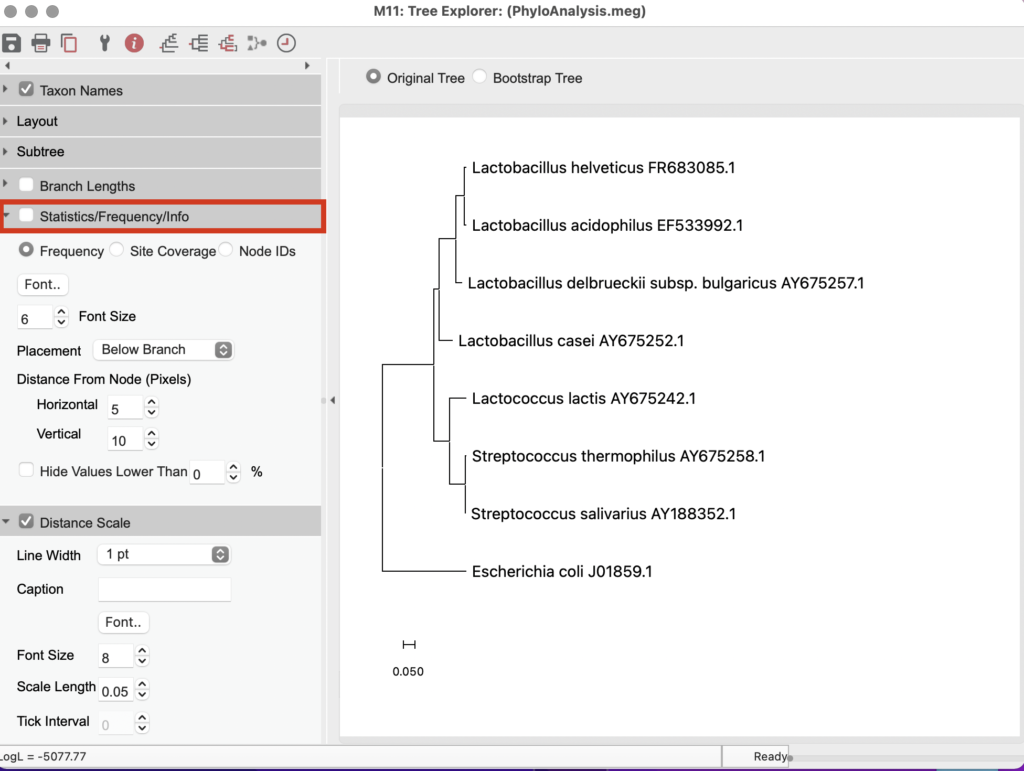
系統樹が構築されました。
BootStrap値を系統樹上で表示するにはサイドメニュー中の
Statics/Frequency/Infoにチェックを入れましょう。
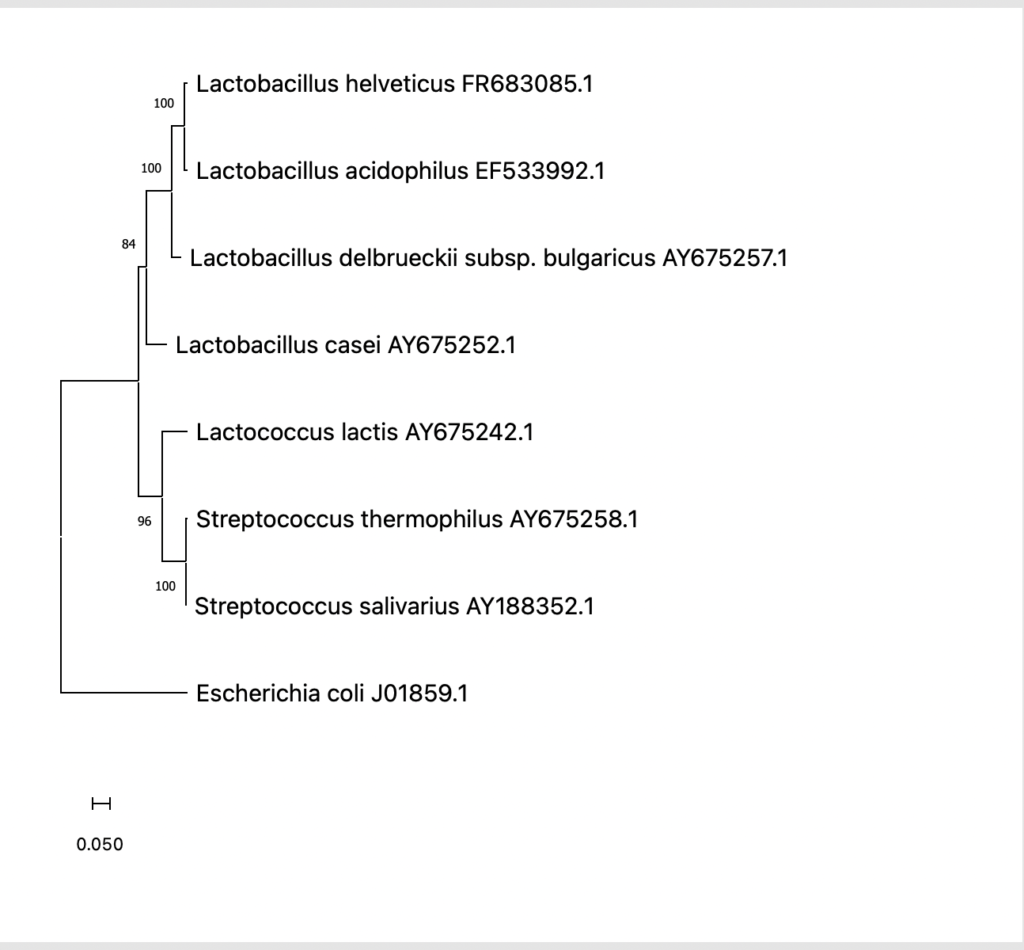
系統樹上にBootStrap値が表示されました。
系統樹を保存する場合は上部の「Image」からfile形式を選択し、保存先を指定します。
まとめ
なかなか順を追ってMEGAを用いた系統樹作成法を説明しているサイトは少ないと感じたので、
今回自分がおこなっている方法を紹介してみました。
系統解析にはバイオインフォマティクスの知識が必要不可欠です。
筆者もこれから勉強していきたいと思っています。
![[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]](https://hbb.afl.rakuten.co.jp/hgb/25b26384.9cbbf86d.25b26385.79ac56b5/?me_id=1213310&item_id=20523856&pc=https%3A%2F%2Fthumbnail.image.rakuten.co.jp%2F%400_mall%2Fbook%2Fcabinet%2F7912%2F9784766427912_1_43.jpg%3F_ex%3D240x240&s=240x240&t=picttext "[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]")

参考文献
- https://ja.wikipedia.org/wiki/16S%E3%83%AA%E3%83%9C%E3%82%BD%E3%83%BC%E3%83%A0RNA
- https://www.sbj.or.jp/wp-content/uploads/file/sbj/9110/9110_yomoyama.pdf